
Flavivirus-GLUE
Comparative Genomics Across Evolutionary Scales



1. Comparative Genomic Analysis of Flaviviruses
The Flaviviridae—particularly the ‘classical’ flaviviruses of the genus Flavivirus—have played a central role in the development of comparative virology, largely because of their importance as human and animal pathogens. Long before genomic approaches became routine, flaviviruses were the subject of sustained investigation through classical virology, immunology, and epidemiology. As a result, flavivirus diversity was relatively well characterised in biological terms.
During the 20th century, extensive sampling driven by human and veterinary disease surveillance produced virus collections that were tied to specific regions, hosts, and transmission cycles. Many flaviviruses identified in this era were isolated, propagated, antigenically typed, associated with particular reservoir hosts or vectors, and linked to characteristic disease phenotypes. This body of work tied flavivirus diversity to real ecological settings, spanning tropical forests, cave systems, river basins, and islands.

This historical foundation matters because much of flavivirus diversity was characterised biologically before genomic sequence data became the primary lens for virus discovery. When molecular phylogenetic and genomic approaches later emerged, existing biological, epidemiological, and ecological knowledge about flaviviruses could therefore be organised and compared in an evolutionary framework.
This comparative, phylogeny-based perspective now underpins how genomic data are interpreted and exploited in flavivirus research. Earlier applications focused largely on patterns of emergence, spread, and vector association, but the same framework increasingly supports a broader range of analyses that connect sequence variation to structure, function, and phenotype across flaviviruses. These draw on advances in protein structural biology and structure prediction, as well as an improved mechanistic understanding of flavivirus replication and transmission.
Such analyses span multiple evolutionary scales: genotype–phenotype relationships, immune escape, and antiviral susceptibility are often examined within species or closely related clades, while conserved replication mechanisms, structural constraints, and functional domains are revealed through deeper comparisons at genus or subgenus levels.
In an era of rapidly expanding virus discovery through metagenomics, exploiting the full range of comparative opportunities requires more than large sequence datasets. It requires a framework in which evolutionary relationships are made explicit, and analyses can be deliberately scoped to the appropriate depth.
Flavivirus-GLUE was designed to meet this requirement. It provides a structured, multi-scale representation of flavivirus diversity via a hierarchy of reference-constrained alignments. The following sections describe the alignment strategy used in Flavivirus-GLUE and show how this structure can be exploited in practical comparative analyses.
2. Alignment Strategy: Multi-Scale, Translation-Informed, Hierarchical

Flavivirus-GLUE is designed to make evolutionary context explicit and operational for comparative genomics. It does this by representing flavivirus diversity through a hierarchically organised set of multiple sequence alignments that capture homology at different evolutionary depths. This strategy allows comparative analyses to be deliberately scoped—within species, across clades, or at deeper taxonomic levels—so that analytical resolution is matched to the biological question
2.1. Translation-informed nucleotide alignments as the backbone
All primary sequence data in Flavivirus-GLUE are stored and aligned as nucleotide sequences. However, because deep evolutionary comparisons at the nucleotide level are often unreliable across the Flaviviridae, alignment construction is guided by translation and protein-level homology.
In practice, this means that coding regions are aligned in nucleotide space, but under constraints derived from their translated amino-acid sequences. Conserved proteins—most notably NS5, the RNA-dependent RNA polymerase—provide a stable framework for establishing positional homology across divergent taxa, while preserving the underlying nucleotide representation required for downstream analyses.
This approach ensures that alignments remain biologically interpretable at both the protein and nucleotide levels.
2.2. Codon-aware representation and feature mapping
Because alignments are maintained in nucleotide space, coding regions retain explicit codon structure throughout the project. Genome features are defined relative to reference sequences and mapped consistently across alignments, allowing codon positions, reading frames, and feature boundaries to be queried directly.
This design supports analyses that depend on nucleotide or codon level information (e.g. calculating dn/ds ratios), while still benefiting from protein-informed alignment constraints. Rather than converting between independent protein and nucleotide MSAs, Flavivirus-GLUE maintains a single, coherent representation.
2.3. Hierarchical alignment structure
A central concept in Flavivirus-GLUE is the alignment tree: a hierarchically organised set of constrained multiple sequence alignments that represent homology at different evolutionary depths.
In GLUE, a constrained alignment is an alignment in which all member sequences are mapped onto the coordinate space of a chosen reference sequence. Insertions relative to the reference are preserved explicitly, so sequence data are never discarded.
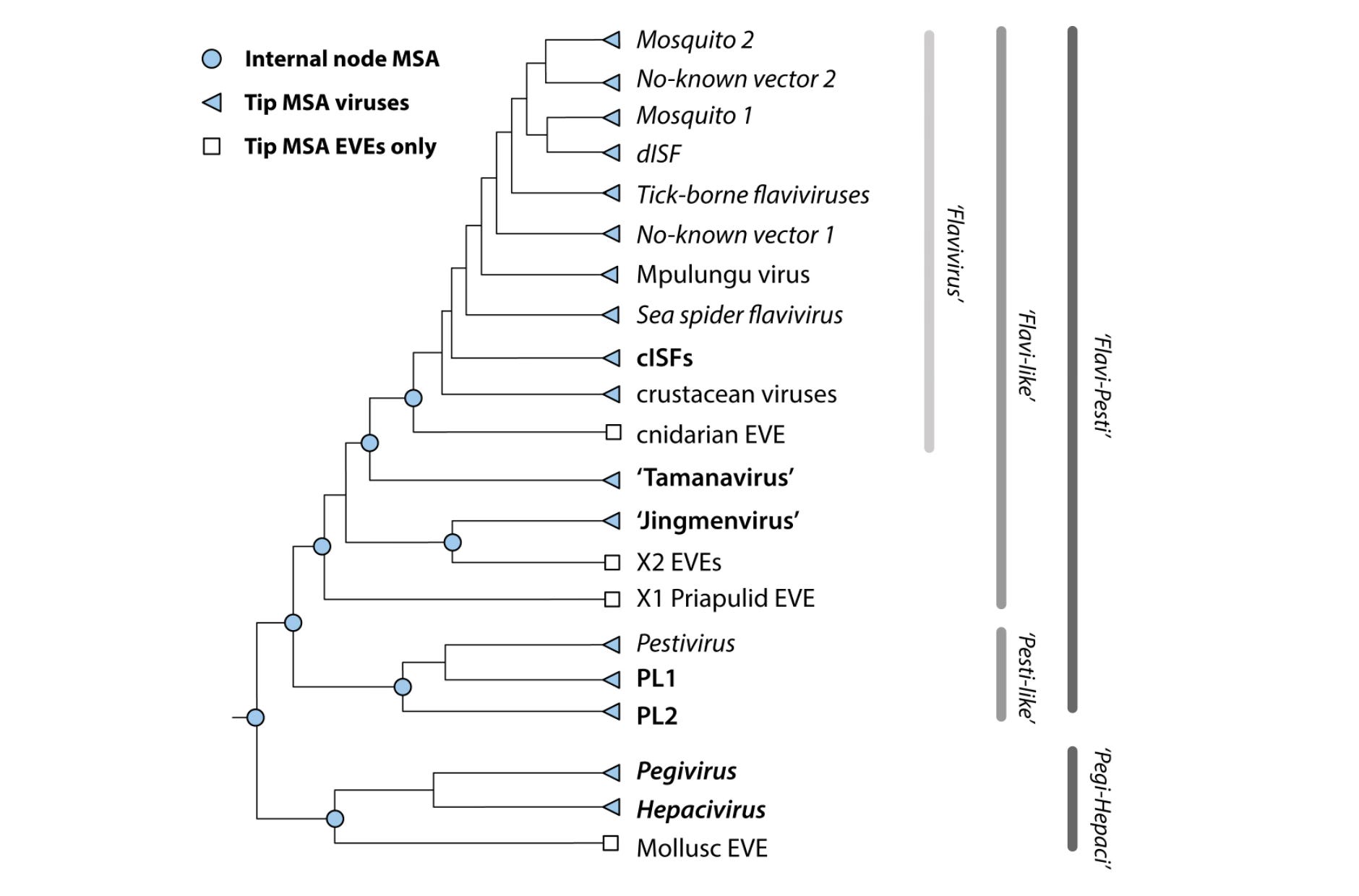
By linking constrained alignments into a tree structure, GLUE allows comparative analyses to be carried out coherently across distinct evolutionary scales. This avoids the need to force all sequences into a single alignment.
family- and major-clade alignments capture deep relationships using conserved regions,
genus-level alignments span as much of the coding genome as homology allows,
subgenus or clade-restricted alignments support finer-scale comparative analyses across complete or near-complete genomes.
3. A Practical Walkthrough Using Flavivirus-GLUE in Docker

This walkthrough illustrates how the Flavivirus-GLUE alignment tree represents evolutionary structure, and how it can be exploited for practical comparative analysis. All examples were run inside a Docker container at the GLUE> prompt.
I installed the Dockerised version Flavivirus-GLUE by following the instructions in the User Guide.
On my M4 Macintosh laptop, the final command in the install series was:
docker run --rm -it \
--platform linux/amd64 \
--name gluetools \
--link gluetools-mysql \
-v "$(pwd):/work" \
-w /work \
cvrbioinformatics/gluetools:latestNote the use of the -v option, which binds the container’s working directory to the local directory, allowing exported files (e.g. alignments) to be written directly to the host file system, as explained in detail here.
Executing the command brings up the interactive GLUE console:
GLUE Version 1.1.113
Copyright (C) 2015-2020 The University of Glasgow
This program comes with ABSOLUTELY NO WARRANTY. This is free software, and you
are welcome to redistribute it under certain conditions. For details see
GNU Affero General Public License v3: http://www.gnu.org/licenses/
Mode path: /
Option load-save-path: /work
GLUE> …which will be used throughout this tutorial.
3.1 Navigating into the Project
We begin by listing available GLUE projects:
Mode path: /
Option load-save-path: /work
GLUE> list project
+==============+===================================+
| name | description |
+==============+===================================+
| flaviviridae | GLUE project for the Flaviviridae |
+==============+===================================+
Projects found: 1Now let’s enter the ‘flaviviridae’ project:
Mode path: /
Option load-save-path: /work
GLUE> project flaviviridae
OKWe can now access project-specific functions and data. Available functions and options can be seen be using the GLUE console’s “tab auto-complete” feature.
GLUE>
alignment commit compute
concatenate config console
copy count create
custom-table-row data-util delete
exit export extend
feature file-util generate
glue-engine help import
list module move
multi-copy multi-delete multi-render
multi-set multi-unset new-context
project-mode quit reference
root-mode run sequence
set show translate
unset validate web-list Type help <command> for any of the displayed options to obtain details. In GLUE, help <command> provides context-specific documentation for the current mode (project, table, module, etc.).
3.2 Inspecting the Alignment Hierarchy

Listing alignments provides an immediate overview of how the project is organised:
GLUE> list alignmentFor each constrained alignment, GLUE reports:
the alignment name,
its parent alignment (if any),
and the reference sequence that defines its coordinate space.
This reflects the hierarchical alignment strategy shown in the schematic in Section 2.
It will differ slightly, however, because the schematic includes nodes associated with endogenous viral elements (EVEs). These are not included in the core project, but can be incorporated via an additional project layer, in line with GLUE’s layered project architecture.
3.3 Exporting Feature-Defined Nucleotide Alignments
GLUE provides the fastaAlignmentExporter module for exporting nucleotide alignments. An instance of this module is included in the distributed build of the Flavivirus-GLUE project. Before using this module to export, it is useful to inspect the available options:
GLUE> module fastaAlignmentExporter help exportAs an example, we can export the NS3 coding region from a subgenus-level alignment of group 1 mosquito-borne flaviviruses (MBFV1). This is the group that includes Dengue virus and Zika virus among many others:
GLUE> module fastaAlignmentExporter \
export AL_FLAVI_SUBGENUS_MBFV1 \
-r REF_MASTER_DEN1 -f NS3 -a -e -p This command exports a nucleotide alignment for the NS3 coding region from the subgenus-level mosquito-borne flavivirus alignment (AL_FLAVI_SUBGENUS_MBFV1). The command options have the following meanings:
-rspecifies the reference sequence that defines the alignment coordinate space,-fselects a genome feature to export (use tab auto-completion to list available features),-aexports all qualifying alignment members,-eexcludes rows with no sequence data for the selected feature,-ppreviews the resulting FASTA output directly at the console.
With the -p option, output is written to the terminal. Supplying -o <filename> instead of -p writes the FASTA file to the project’s load/save directory.
Note: For readability, the backslash (\) character is used above to split the command across multiple lines; when entering commands at the GLUE prompt, they should be supplied on a single line.
The command shown above is equivalent to the following two-step sequence:
Mode path: /project/flaviviridae
GLUE> module fastaAlignmentExporter
OK
Mode path: /project/flaviviridae/module/fastaAlignmentExporter
GLUE> export AL_FLAVI_SUBGENUS_MBFV1 -r REF_MASTER_DEN1 -f NS3 -a -e -pNotice that the mode path is updated when we navigate into the module, indicating that subsequent commands are now evaluated in the context of that module. In this mode, the export command is available directly without further qualification.
In the examples that follow, we will generally enter the relevant module explicitly so that command statements are shorter and easier to read, although issuing fully qualified commands from project mode remains equally valid.
GLUE provides several commands for navigating back up through the data model hierarchy. The exit command moves up one level at a time, returning from the current command mode to its parent context. Alternatively, the project-mode command can be used to return directly to the top-level project context from any deeper mode.
3.4 Exporting Protein Alignments

Protein alignments are exported using a separate module type. For example, we can export a protein alignment for NS1 from the same subgenus-level alignment as we used for our nucleotide example:
Mode path: /project/flaviviridae/module/fastaProteinAlignmentExporter
GLUE> export AL_FLAVI_SUBGENUS_MBFV1 -r REF_MASTER_DEN1 -f NS1 -a -e -p3.5 Working Across the Alignment Tree
The alignment tree becomes particularly powerful when exporting alignments at different levels of the hierarchy. The examples above operated at the tips of the alignment tree. Tip alignments correspond to relatively narrow evolutionary groupings with high sequence coverage.
For example, the following command exports an NS5 protein alignment from a subgenus-level alignment of tick-borne flaviviruses:
Mode path: /project/flaviviridae/module/fastaProteinAlignmentExporter
GLUE> export AL_FLAVI_SUBGENUS_TBFV -r REF_MASTER_POWV -f NS5 -a -e -pThis produces a feature-aligned protein alignment representing NS5 across tick-borne flaviviruses.
The same operation can be performed at a higher level of the alignment tree, such as the genus-level Flavivirus alignment:
Mode path: /project/flaviviridae/module/fastaProteinAlignmentExporter
GLUE> export AL_GENUS_Flavivirus -r REF_MASTER_YFV -f NS5 -a -e -pAlthough the same feature is exported, the coordinate system is now defined by a genus-level reference sequence. Internal node alignments act as structural hubs that link multiple descendant clades through shared reference sequences.
By default, exporting from an internal node returns only the sequences directly associated with that alignment (i.e. the constraining reference sequences inherited from its child alignments). To include all members from all descendant alignments beneath the specified node, the -c (recursive) option can be used:
Mode path: /project/flaviviridae/module/fastaProteinAlignmentExporter
GLUE> export AL_GENUS_Flavivirus -r REF_MASTER_YFV -f NS5 -caep This single command exports a protein alignment that spans all species in genus Flavivirus.
Importantly, no manual merging of FASTA files is required: evolutionary scope is controlled simply by choosing where in the alignment tree to operate.
3.6 A Simple Comparative Query: Amino-Acid Frequencies

Once alignments are in place, GLUE supports lightweight comparative queries that can be applied at different levels of the alignment hierarchy. As a simple example, we can examine amino-acid frequencies at specific codon positions within the NS5 protein.
We begin by issuing an amino-acid frequency query in a subgenus-level alignment corresponding to group 1 mosquito-borne flaviviruses (MBFV1). At this level, the analysis is restricted to a relatively narrow clade with dense sampling.
Mode path: /project/flaviviridae/alignment/AL_FLAVI_SUBGENUS_MBFV1
GLUE> amino-acid frequency -r REF_MASTER_YFV -f NS5 -l 100 102This command reports the distribution of amino acids at each codon position in the specified range, providing a concise summary of conservation and variability within this clade:
+=========+=======+===========+============+============+
| feature | codon | aminoAcid | numMembers | pctMembers |
+=========+=======+===========+============+============+
| NS5 | 100 | V | 35 | 100.00 |
| NS5 | 101 | A | 1 | 2.86 |
| NS5 | 101 | K | 16 | 45.71 |
| NS5 | 101 | Q | 1 | 2.86 |
| NS5 | 101 | R | 17 | 48.57 |
| NS5 | 102 | A | 1 | 2.86 |
| NS5 | 102 | G | 34 | 97.14 |
+=========+=======+===========+============+============+We can perform the same analysis at a higher level of the alignment tree by moving to the Flavivirus genus-level alignment and adding the -c (recursive) option. This instructs GLUE to include all descendant alignments beneath the genus-level node.
Mode path: /project/flaviviridae/alignment/AL_GENUS_Flavivirus
GLUE> amino-acid frequency -r REF_MASTER_YFV -f NS5 -l 100 102 -cThe resulting output now reflects amino-acid frequencies across a much broader evolutionary scope:
+=========+=======+===========+============+============+
| feature | codon | aminoAcid | numMembers | pctMembers |
+=========+=======+===========+============+============+
| NS5 | 100 | A | 2 | 1.83 |
| NS5 | 100 | I | 2 | 1.83 |
| NS5 | 100 | V | 105 | 96.33 |
| NS5 | 101 | A | 1 | 0.92 |
| NS5 | 101 | D | 1 | 0.92 |
| NS5 | 101 | E | 2 | 1.83 |
| NS5 | 101 | H | 3 | 2.75 |
| NS5 | 101 | K | 39 | 35.78 |
| NS5 | 101 | L | 9 | 8.26 |
| NS5 | 101 | M | 2 | 1.83 |
| NS5 | 101 | N | 7 | 6.42 |
| NS5 | 101 | Q | 2 | 1.83 |
| NS5 | 101 | R | 36 | 33.03 |
| NS5 | 101 | S | 1 | 0.92 |
| NS5 | 101 | T | 2 | 1.83 |
| NS5 | 101 | V | 2 | 1.83 |
| NS5 | 101 | Y | 2 | 1.83 |
| NS5 | 102 | A | 38 | 34.86 |
| NS5 | 102 | G | 68 | 62.39 |
| NS5 | 102 | M | 1 | 0.92 |
| NS5 | 102 | S | 2 | 1.83 |
+=========+=======+===========+============+============+By simply changing the alignment context and enabling recursive traversal of the alignment tree, the same comparative query can be applied at different evolutionary depths. This illustrates how Flavivirus-GLUE allows comparative genomics analyses to be scaled naturally—from clade-restricted comparisons to genus-wide summaries—without restructuring datasets or manually merging results.
4. Summary and Outlook

Comparative genomics has become central to how flavivirus diversity is interpreted, building on a long tradition of biologically grounded virus research. As genomic data are increasingly used to connect sequence variation to viral structure, function, and phenotype, analyses routinely span multiple evolutionary scales—from within-species comparisons to deep, family-level perspectives. Realising the full value of these approaches requires a framework in which evolutionary relationships are explicit and analytical scope can be matched deliberately to biological questions.
Flavivirus-GLUE was designed to meet this need by representing flavivirus diversity through a hierarchically organised set of reference-constrained alignments. As illustrated in this tutorial, this structure allows comparative analyses to be scaled naturally by operating at different points in the alignment tree, without manual data restructuring. In an era of rapid virus discovery and expanding sequence databases, such explicitly evolutionary, multi-scale representations provide a practical foundation for integrating new data with existing biological knowledge and for maintaining interpretability as datasets continue to grow.
 | A guest post by
|